Overview
Thalassaemia refers to a group of disorders characterised by reduced or absent globin chain production.
Thalassaemia is one of the haemoglobinopathies, which refers to a group of autosomal recessive inherited disorders that affect the globin chains that form the protein component of haemoglobin.
Haemoglobinopathies
Haemglobinopathies can be broadly divided into two types:
- Haemoglobin variants: mutant forms of haemoglobin that affect the structure. Sickle cell disease is most well recognised.
- Thalassaemia: reduced or absent globin chain production due to underlying mutations.
Thalassaemia
Thalassaemia can be further divided depending on the type of globin chain affected:
- Alpha thalassaemia: reduced or absent production of the alpha globin chains
- Beta thalassaemia: reduced or absent production of the beta globin chains
Haemoglobin
Haemoglobin is the main oxygen-carrying molecules within our red blood cells.
Haemoglobin (Hb) is essential for the transport of oxygen around the body. It is composed of four globin chains and four heme molecules, which are the actual oxygen-binding structures that contain iron.
Type of haemoglobin
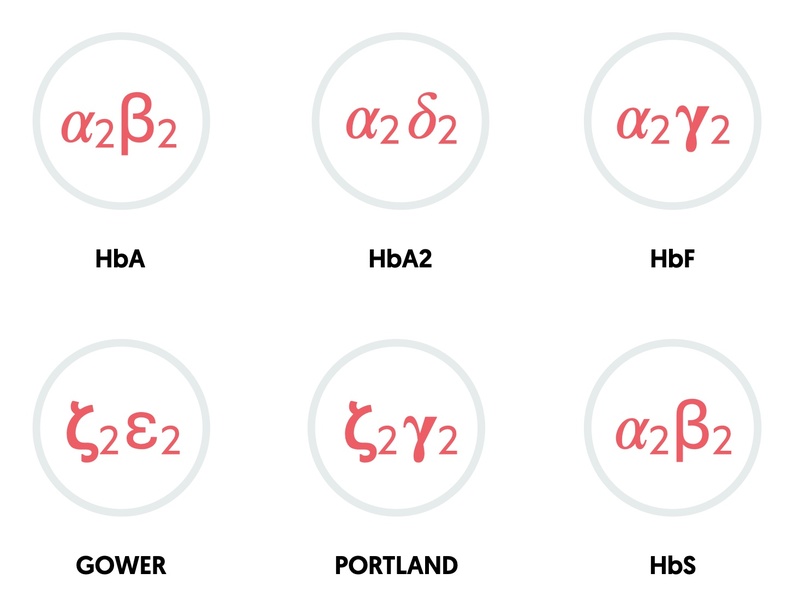
The combination of different globin chains determines the type of haemoglobin.
- HbA: two alpha, two beta (95-98% in adults)
- HbA2: two alpha, two delta (2-4% in adults)
- HbF: two alpha, two gamma (fetal haemoglobin: 0.8-2% in adults)
- Gower: two zeta, two epsilon (embryonic haemoglobin)
- Portland: two zeta, two gamma (embryonic haemoglobin)
Genetics
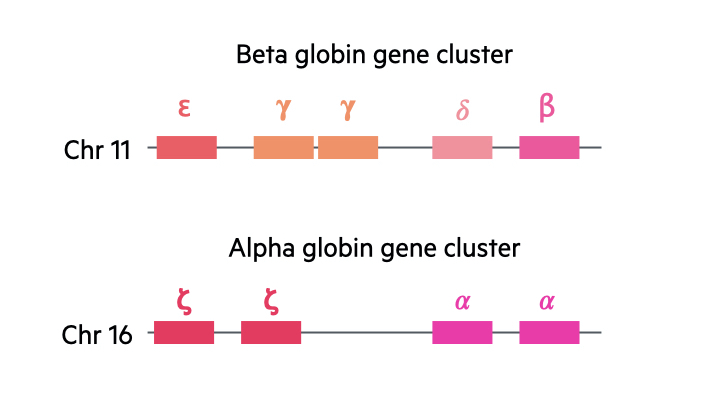
In the human body, there are two major gene clusters important for the synthesis of globin chains in the formation of haemoglobin:
- Alpha globin gene cluster: located on chromosome 16. Contains the embryonic globin genes zeta and two copies of the alpha globin gene (alpha-1 and alpha-2).
- Beta globin gene cluster: located on chromosome 11. Contains the embryonic globin gene epsilon, fetal globin genes and the adult beta globin and delta globin gene.
We contain four alpha globin genes and two beta globin genes. With the alpha genes, two are inherited paternally and two inherited maternally. With the beta globin genes, one is inherited from each parent.
Mutations types
When we look at abnormal mutations seen in thalassaemia, we use the following denotation:
- Mutation leading to absent production (0)
- Mutation leading to reduced production (+)
Epidemiology
Alpha thalassaemia is one of the most common single gene disorders worldwide.
Anyone may be a carrier of an alpha thalassaemia gene, but is it most prevalent in sub-Saharan Africa, the Middle East, Mediterranean regions and areas of Asia (South and South-East).
Globally, it is estimated that 3 in 100 of the world’s population will have a thalassaemia gene (alpha or beta).
Aetiology
In alpha thalassaemia, mutations in the alpha globin genes lead to reduced or absent production.
We each have four alpha globin genes. The severity of alpha thalassaemia therefore depends on how many genes are affected and whether the mutations leads to reduced, or absent, production. In general, there are two clinically significant forms of alpha thalassaemia that occur if three or four of the genes are affected.
Genetic mutations
Numerous genetic mutations may occur in the alpha globin genes leading to thalassaemia. Deletions of the alpha globin genes are the most commonly seen mechanisms accounting for alpha thalassaemia, particularly in Asian and Mediterranean populations.
Mutations seen can include:
- Deletions
- Genetic rearrangements
- Mutations affecting transcription (due to mutations that affect promotors or enhancers)
- Mutations affecting splicing (splicing is form of mRNA processing that removes introns)
- Premature stop codons (affects mRNA translation leading to truncated proteins)
- Other rarer mechanisms
Alpha thalassaemia types
Alpha thalassaemia can be considered an autosomal recessive disorder. However, because there are two genes on each chromosome that encode the alpha globin chains, there are multiple types of alpha thalassaemia.
- Inheritance of three normal copies (aa/a-): known as alpha thalassaemia minima. Patients are asymptomatic.
- Inheritance of two normal copies (a-/a- OR aa/–): known as alpha thalassaemia trait. Minimal anaemia.
- Inheritance of one normal copy (a-/–): known as haemoglobin H disease (HbH). HbH is composed of tetramers of beta globin chains. Moderate to severe haemolytic anaemia.
- Inheritance of no normal copies (–/–): known as Hb Barts syndrome. Incompatible with extrauterine life.
Pathophysiology
In the absence of alpha globin chains, normal haemoglobin A, A2 and fetal haemoglobin cannot be formed.
The severity of thalassaemia depends on both the number, and how, the genes are affected.
Alpha thalassaemia trait
Patients with alpha thalassaemia trait are usually asymptomatic. There may be evidence of mild microcytic anaemia on routine blood testing. There are no specific treatments required. The condition is clinically relevant for offspring. If two patients with alpha thalassaemia trait or minima have a child, there is a risk of HbH or Hb Barts syndrome.
Haemoglobin H (HbH) disease
Patients with HbH disease inherit three abnormal copies of the alpha globin chain. These may be deletional or non-functional mutations. Without alpha globin chains, there are two major consequences:
- Neonatal period: formation of abnormal haemoglobin composed of four gamma chains. Known as Hb Barts.
- Adult period: formation of abnormal haemoglobin composed of four beta chains. Known as HbH.
The consequence of the abnormal HbH is a reduced red cell survival with a hypochromic microcytic anaemia. These cells are also removed from the circulation at an increased rate by the spleen. The severity of anaemia is variable depending on the mutation.
The HbH haemoglobin is at increased risk of oxidative stress and is ineffective as an oxygen delivering molecule. This is because, like myoglobin, it holds onto oxygen with higher affinity. It is estimated to have an affinity for oxygen at least 10 times that of HbA (normal haemoglobin).
Haemoglobin Barts
This is the most severe form of alpha thalassaemia that results from inheritance of four abnormal copies of the alpha globin gene. The condition is incompatible with extrauterine life.
It is characterised by the formation of Hb Barts, which is composed of four gamma globin chains. Normal haemoglobin synthesis begins in the 5-6th weeks of intrauterine life, so effects are seen early in pregnancy. Like HbH, Hb Bart’s binds to oxygen with high affinity and is unable to release it to tissues.
Due to the presence of embryonic globin chains, the foetus can survive into the third trimester. However, the poor oxygen delivery, reduced red blood cell survival and compensatory extramedullary erythropoiesis leads to severe anaemia, high-output cardiac failure and generalised oedema. This clinical picture is known as Hydrops fetalis.
Clinical manifestations
The clinical spectrum of alpha thalassaemia is highly variable depending on the type and number of mutations.
Alpha thalassaemia minima
- Asymptomatic
- No evidence of anaemia
- Normal mean corpuscular volume (MCV)
Alpha thalassaemia trait
- Asymptomatic
- Normal or mild anaemia
- Usually mild reduction in MCV
HbH
The clinical picture of HbH is highly variable. Some patients will be relatively asymptomatic with mild degrees of anaemia. Other patients may require intermittent transfusions, whereas others have severe anaemia with marked extramedullary haematopoiesis.
- Pallor
- Anaemia
- Jaundice (unconjugated due to chronic haemolytic anaemia)
- Gallstone disease (chronic haemolysis)
- Extramedullary haematopoiesis: hepatosplenomegaly, skeletal changes
- Osteopenia/osteoporosis
- Aplastic/hypoplastic crisis: sudden reduction in red blood cell counts
- Leg ulcers
Hb Barts
Hb Barts is incompatible with extrauterine life. There are reports of patients surviving for variable periods following birth, but this is with red blood cell transfusions and advanced medical care.
Diagnosis & investigations
The majority of patients with alpha thalassaemia will be completely asymptomatic.
Most patients with alpha thalassaemia minima or minor will be completely asymptomatic and not realise they have the underlying condition. More severe forms of alpha thalassaemia may be suspected in babies or children presenting with anaemia and jaundice.
Initial testing
- Full blood count: looking for low haemoglobin and low mean corpuscular volume (MCV)
- Blood film: typically shows hypochromic, microcytic red blood cells. Abnormal red cell morphology is characteristic of HbH
- Liver function tests: unconjugated hyperbilirubinaemia
- Haemolysis screen: Lactate dehydrogenase (raised), haptoglobin (reduced), direct antiglobulin test (Negative, non-immune haemolysis)
- Iron studies: iron deficiency is a major cause of microcytic anaemia that needs to be excluded. Elevated iron seen if high turnover of red blood cells.
Diagnostic testing
Babies presenting within 6 months of birth are more likely to have alpha thalassaemia because the alpha globin chain is already needed in production of haemoglobin.
- Haemoglobin analysis: completed using haemoglobin electrophoresis or high-performance liquid chromatography (HPLC). Electrophoresis causes different types of haemoglobin to separate into bands. HPLC is an alternative method of determining the types of haemoglobin in blood.
- Genetic testing: DNA testing provides a definite and precise diagnosis of alpha thalassaemia. Importantly, genetic testing is the only method of detecting alpha thalassaemia minima or trait.
On haemoglobin analysis, patients with Hb Barts will have a complete absence of HbA, HbA2 and HbF because of an inability to produce alpha globin chains. Patients with HbH will have clear evidence of HbH alongside elevated levels of HbF and HbA2. There may be small amounts of Hb Barts.
Antenatal screening
Screening for thalassaemia is offered to all pregnant women within the UK.
Antenatal screening is offered to all pregnant women within the UK. It involves concurrent assessment of different haemoglobinopathies (i.e. thalassaemia and haemoglobin variants) at 10 weeks gestation. Family origin questionnaires are essential as part of the screening programme to determine the risk of being a carrying of a haemoglobinopathy gene.
In beta thalassaemia, the level of HbA2 is quantified. Levels of HbA2 >3.5% is suggestive of being a beta thalassaemia carrier and further analysis of the father is required to determine the risk of beta thalassaemia in the fetus.
Screening for alpha thalassaemia is more difficult. This is because detection of alpha thalassaemia minima (aa/a-) or alpha thalassaemia trait (a-/a-) can only be completed by DNA testing as there are no specific biomarkers. Therefore, in pregnant mothers if the mean corpuscular haemoglobin is < 25 pg and they are from a high risk area (e.g. China, Southeast asian, etc) then testing of the biological father should be offered. If the biological father is also suspected of having alpha thalassaemia then both the mother and biological father should undergo DNA analysis.
Public Health England (PHE) have produced clear guidelines on the sickle cell and thalassaemia screening programme.
Management
Patients with alpha thalassaemia minima or trait do not require treatment.
There is no specific treatment for patients with mild forms of alpha thalassaemia. The only issue is related to antenatal care if the unborn child is at risk of more severe forms of alpha thalassaemia. There are novel treatments for patients with Hb Barts disease including intrauterine blood transfusions and bone marrow transplants, but this is extremely specialist and not discussed here.
HbH disease
Management in alpha thalassaemia focuses on patients with HbH disease, which has a wide spectrum of severity. Some patients may require sporadic transfusions, whereas others remain transfusion dependent due to chronic, severe haemolytic anaemia. Some of the key principles of management are discussed below:
- Transfusions: Haemoglobin typically ranges from 70-100 g/L. Transfusions may be needed for patients with Hb <70 g/L. Local protocols should be followed in association with haematology.
- Iron overload: Patients undergoing regular transfusions are at risk of iron overload and secondary haemochromatosis. They usually require iron chelation therapy (e.g. Deferasirox).
- Dietary supplementation: folic acid is usually recommended in patients with chronic haemolysis.
- Splenectomy: may be considered to reduce transfusion requirements and hypersplenism (reduces number of circulating blood cells).

