Overview
The syndrome of inappropriate anti-diuretic hormone (SIADH) results from excess ADH secretion.
ADH excess, as the name suggests, results in reduced diuresis – water excretion and urinary output are reduced. This leads to an increase in total body water and hyponatraemia.
The condition is common and has an enormous number of causes. Once recognised fluid restriction is the mainstay of management with careful monitoring of plasma sodium levels.
NOTE: Osmolality is a measure of osmoles of solute per kilogram of solvent (Osm/kg).
Physiology
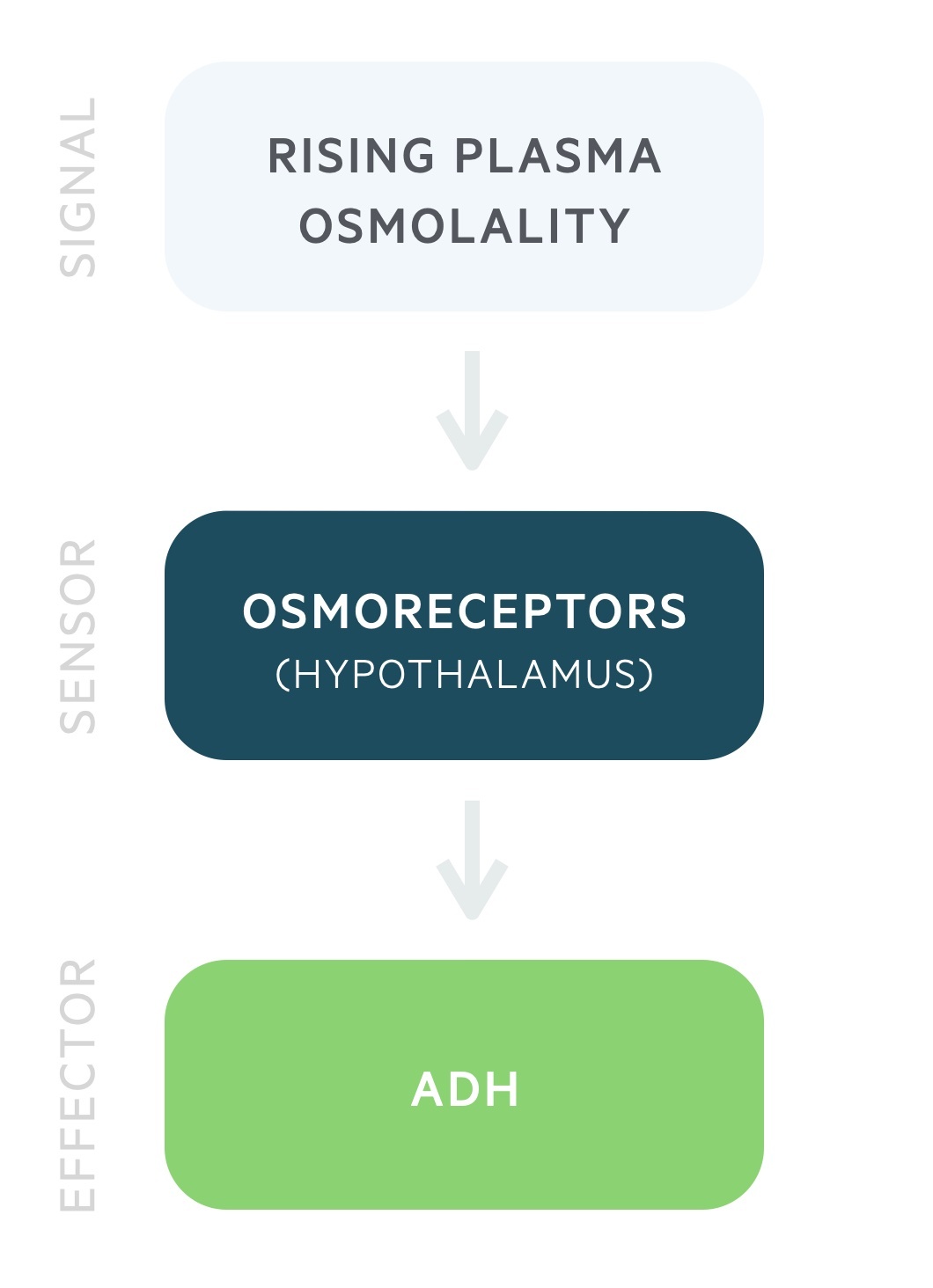
ADH release is governed by the plasma osmolality.
Antidiuretic hormone (ADH) is produced by the magnocellular neurons in the paraventricular and supraoptic nuclei of the hypothalamus. It is stored and released by the posterior pituitary in response to rising plasma osmolality. It may also be referred to as arginine vasopressin (AVP) or simply vasopressin.
ADH release begins at a plasma osmolality of around 280 mOsm/kg. Thirst is felt at around 290 mOsm/kg. These processes lead to a reduction in the plasma osmolality.
ADH acts on the distal convoluted tubule and collecting duct to increase water reabsorption independent of sodium. ADH stimulates the insertion of aquaporin-2 channels onto the luminal membrane, allowing the free entry of water. ADH also causes vasoconstriction of arterioles.
Pathophysiology
SIADH results in a hypotonic hyponatraemia and concentrated urine.
ADH release normally increases with rising plasma osmolality leading to a reduction in water excretion. This results in a fall in the plasma osmolality. The pathophysiology of SIADH is complicated and there are ongoing debates as to how different mechanisms may interact.
In SIADH abnormal release results in the kidneys retaining more water leading to an increase in total body water (TBW). This leads to a dilutional hyponatraemia. Though SIADH is classically described as a euvolaemic hyponatraemia there is some expansion of blood volume. This is evidenced by suppression of renin and an increase in the natriuretic peptides.
The increase in TBW also causes an increase in the extracellular fluid volume that results in increased urinary sodium excretion. This process further contributes to the hyponatraemia whilst helping to return the extracellular fluid volume to normal.
SIADH results in:
- Hypotonic hyponatraemia
- Concentrated urine
- Euvolaemic state
Aetiology
A variety of conditions may predispose a patient to developing SIADH.
The list below is not exhaustive but illustrates how broad the aetiology can be.
Central nervous system
- Stroke
- Subarachnoid haemorrhage
- Trauma
- Psychosis
Neoplasia
- Small cell lung cancer (paraneoplastic syndrome)
- Other lung cancers (less commonly)
- Neuroblastoma
Infective
- Pneumonia
- HIV
Drugs
- Carbamazepine
- Cyclophosphamide
- Selective serotonin reuptake inhibitors (SSRIs)
- MDMA (Ecstasy)
Hormonal
- Hypothyroidism
- Hypopituitarism
Surgery
- Common following many procedures
- Particular risk in transsphenoidal pituitary surgery
Clinical features
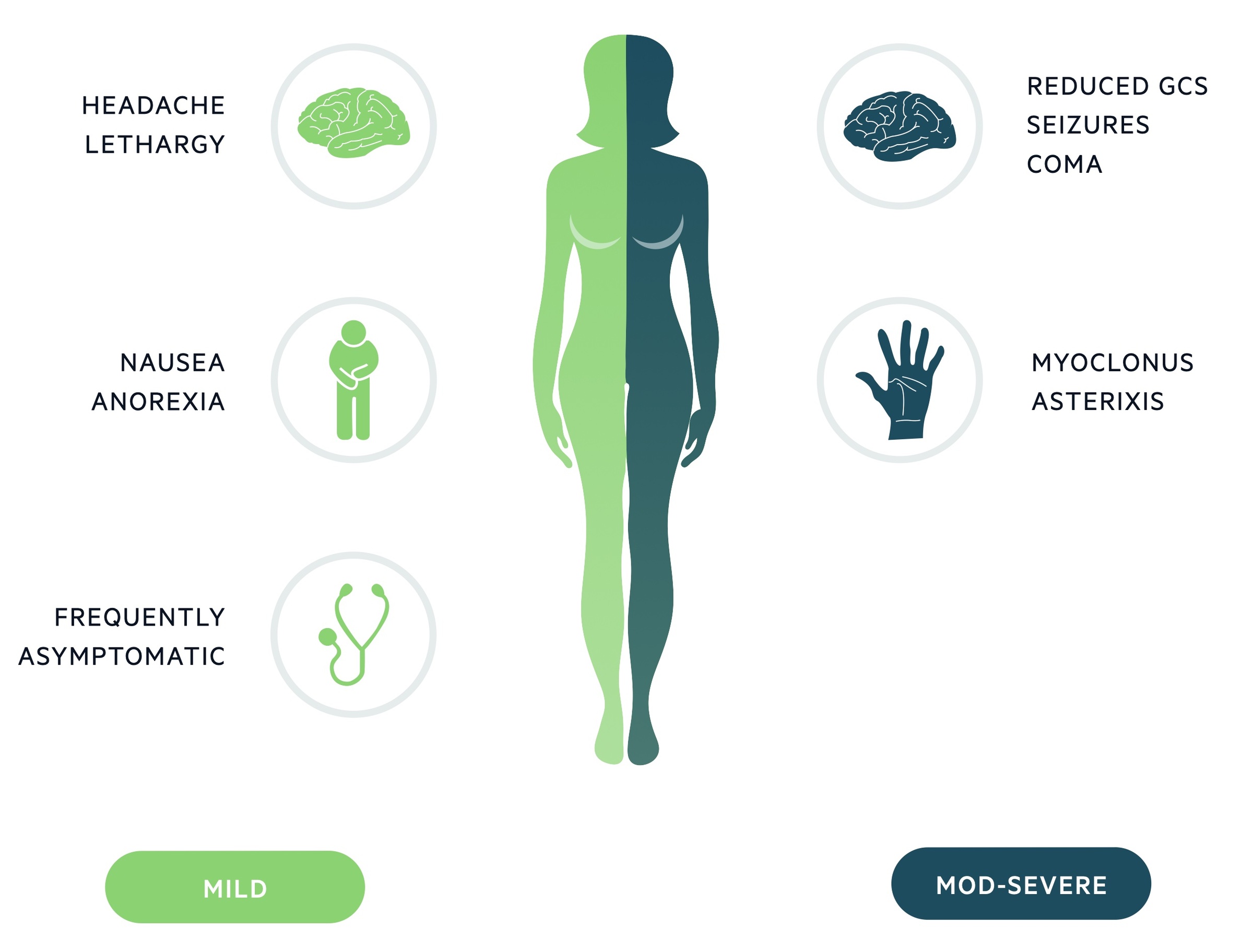
The clinical features associated with SIADH are those of hyponatraemia.
A large proportion of cases will be asymptomatic with clinical features developing with either severe hyponatraemia or following a rapid fall in the sodium level. Neurological signs will only be seen in severe cases.
The clinical features may also reflect the underlying cause. As you can surmise from the Aetiology chapter above we cannot go into each of these in detail.
Symptoms
- Headache
- Confusion
- Lethargy
- Anorexia
Signs
- Seizures
- Reduced GCS
- Coma
- Myoclonus
- Ataxia
- Hyporeflexia
- Asterixis
Diagnosis
SIADH is suspected based on results of clinical chemistry and the patients history.
There is no single test that conclusively diagnoses SIADH. Instead, diagnosis relies upon suggestive biochemistry results and the clinical context.
Blood tests
Renal function: gives a serum sodium that confirms hyponatraemia. May also show a low serum urea consistent with mild volume expansion.
Serum osmolality: a low serum osmolality is seen, less than 280 mOsm/kg.
Urinary tests
Urinary osmolality: a high urine osmalality is seen, greater than 100 mOsm/kg.
Urine sodium: typically a high urinary sodium is seen, greater than >40 mmol/L.

Management
Mild disease requires no specific treatment but the underlying cause should be identified and treated. More severe disease requires correction of the hyponatraemia.
Correction of hyponatraemia should not occur rapidly (no faster than 8-10 mmol/L per 24 hours). Rapid increases in plasma sodium may result in osmotic demyelination syndrome.
Fluid restriction
The typical fluid restriction regime is 500 – 1000mL / 24hrs.
Fluid restriction may not be used in SIADH secondary to subarachnoid haemorrhage as it risks lowering blood pressure which may cause cerebral vasospasm and infarction.
Pharmacological
Demeclocycline, a tetracycline antibiotic, causes an iatrogenic nephrogenic diabetes insipidus through the inhibition of ADH. Though historically popular it is rarely used today.
A newer class of drugs termed vaptans (e.g. tolvaptan) are now more commonly used. These vasopressin receptor antagonists are effective but expensive agents.
Severe hyponatraemia
Those with severe hyponatraemia are at risk of permanent neurological damage due to cerebral oedema, typically seen following a rapid (< 48 hrs) drop in sodium to below 120 mmol/L.
Severe hyponatraemia requires intensive care. Hypertonic saline and furosemide may be utilised. Within the intensive care setting a more rapid increase in serum sodium (4-6 mmol/L in the first hour) may be justified.
Long-term management
In many cases, SIADH is self-limiting or reversible (e.g. post-operative, drugs). In others, it may persist for longer periods and require further fluid restriction and oral salt (causes increased diuresis).
Osmotic demyelination syndrome
Osmotic demyelination syndrome, also termed cerebral pontine myelinolysis, is caused by rapid correction of hyponatraemia.
This syndrome occurs due to a rapid increase in extracellular osmolality. It can cause irreversible neurological damage. As such, in the absence of severe neurological features, sodium correction should not exceed 8-10 mmol/L per 24 hours.

