Introduction
Multiple endocrine neoplasia (MEN) syndromes are autosomal dominantly inherited conditions characterised by tumours of endocrine glands.
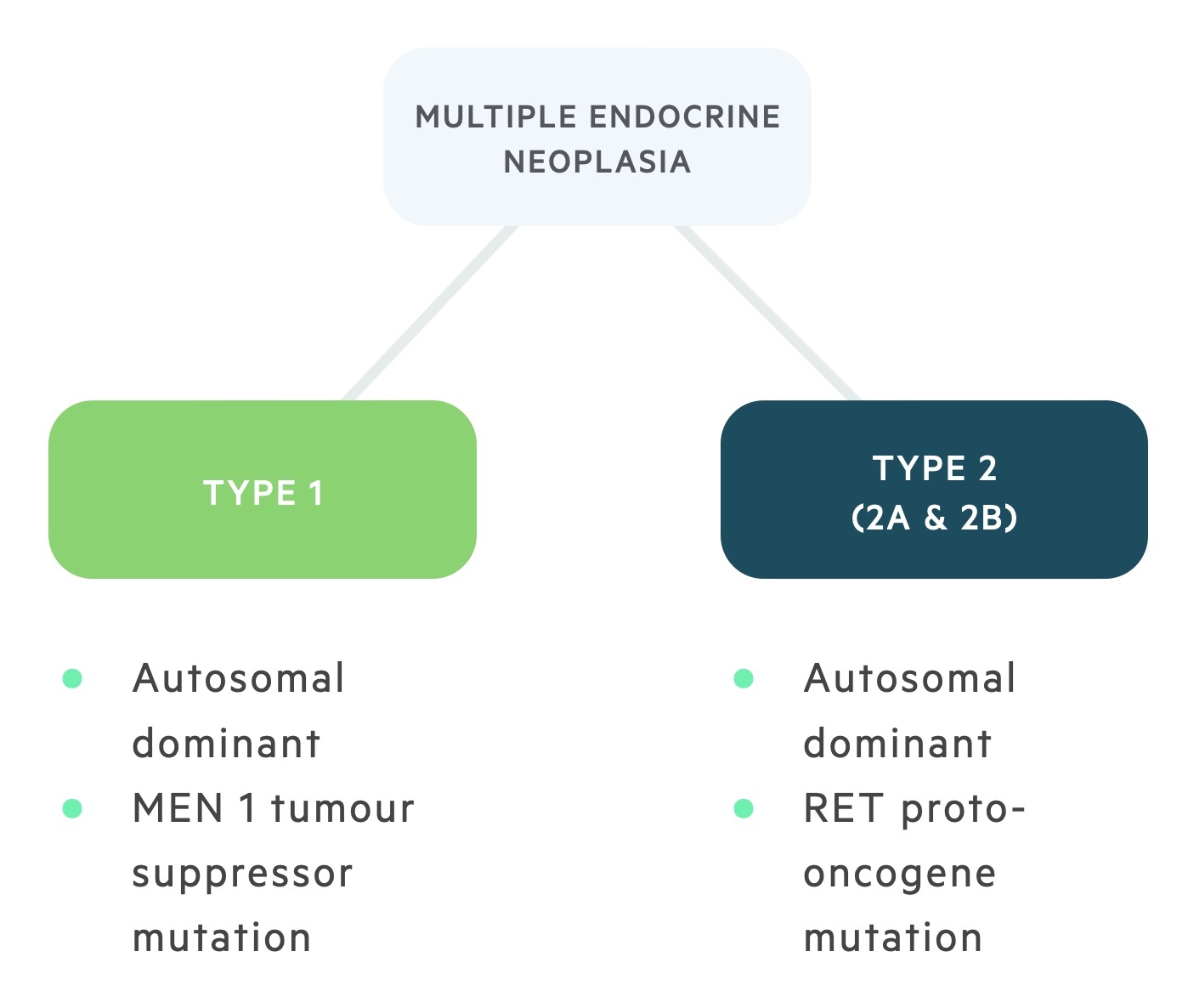
Multiple endocrine neoplasia (MEN) type 1 and 2 are rare autosomal dominant disorders characterised by benign and malignant changes in multiple endocrine glands:
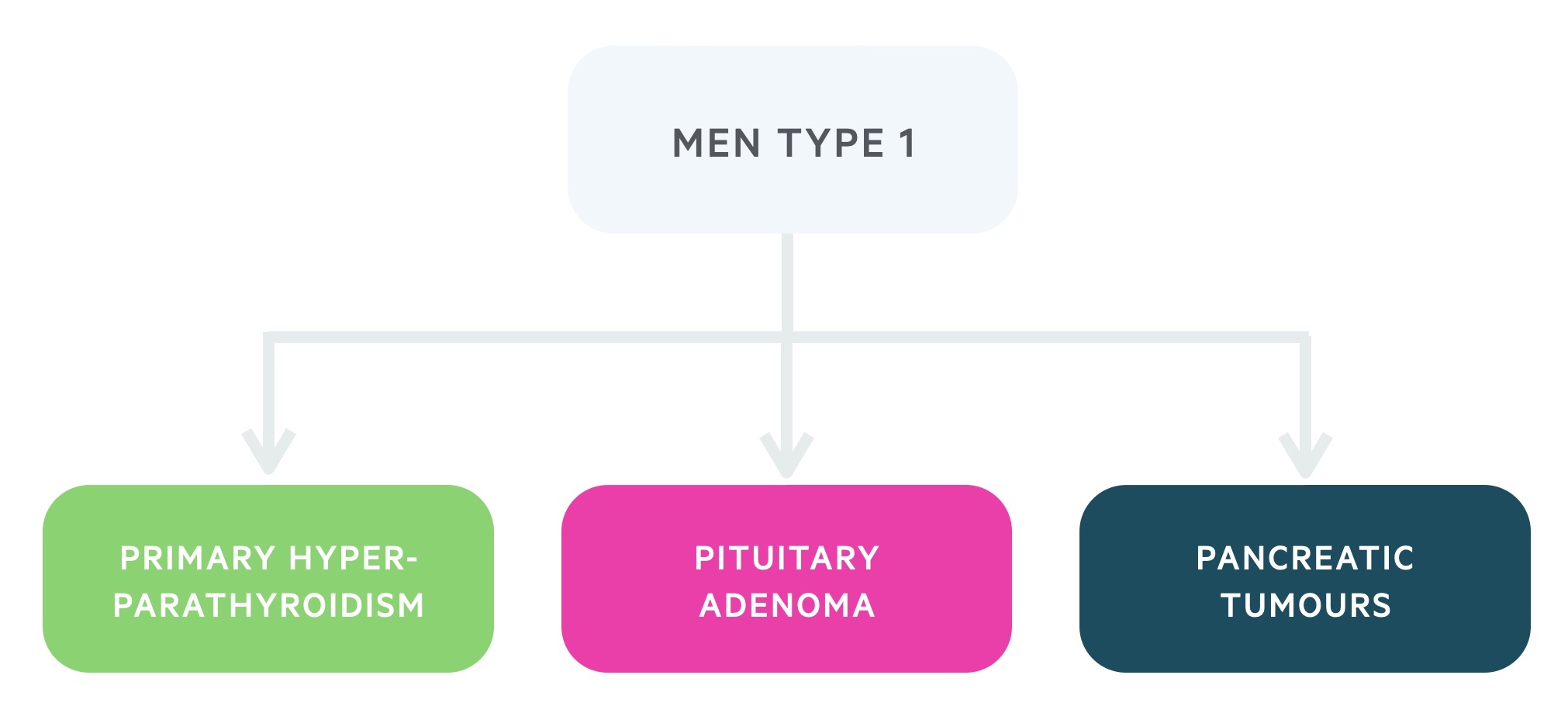
- MEN type 1: is caused by a mutation to the MEN 1 tumour suppressor gene and is characterised by the three P’s; primary hyperparathyroidism, pituitary adenoma and pancreatic tumours.
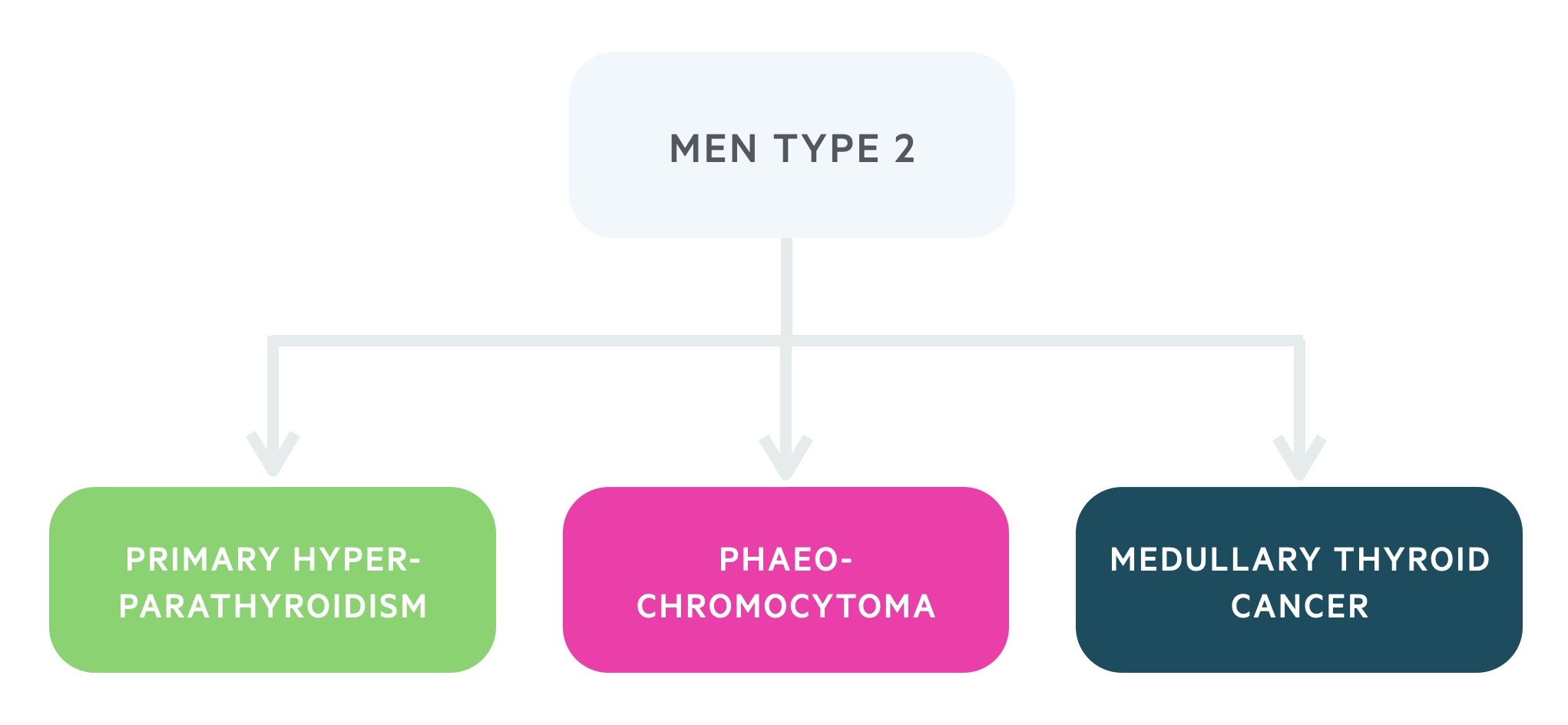
- MEN type 2: is caused by a mutation of the RET proto-oncogene, it is characterised by primary hyperparathyroidism (less common and milder than in type 1), phaeochromocytomas and medullary thyroid cancer. It is divided into two types, MEN 2A (normal appearance, primary hyperparathyroidism is common) and MEN 2B (marfanoid habitus, primary hyperparathyroidism is rare).
MEN type 1
MEN 1 is an autosomal dominant condition with a frequency of 1 in 30,000.
It is caused by a mutation to the MEN 1 tumour suppressor gene. The condition is characterised by a specific collection of endocrine tumours (the three P’s). In addition to the three P’s, cutaneous tumours such as angiofibromas are common.
1. Primary hyperparathyroidism
This occurs in over 90% of patients with MEN 1. It differs from sporadic cases as it presents earlier and has an even sex distribution. Disease usually affects many, if not all, the parathyroid glands and has high rates of recurrence following surgical excision.
Parathyroid hormone has multiple effects on calcium homeostasis including the activation of vitamin D, increasing serum calcium levels. Patients may present with symptoms of hypercalcaemia.
For more information, see our notes on Primary hyperparathyroidism.
2. Pituitary adenomas
These affect around 50% of patients. They can affect a number of different cell types. They are more likely to be macroadenomas (and therefore have features of mass effects) than in sporadic cases of pituitary adenomas.
A number of cell lines may be affected:
- Mammotrophs: Results in hyperprolactinaemia caused by a prolactinoma.
- Somatotrophs: Acromegaly occurs due to excess growth hormone.
- Corticotrophs: Results in the release of excess cortisol (Cushing’s syndrome) through excess production of ACTH.
3. Pancreatic tumours
The two major pancreatic tumours that occur in MEN 1 are insulinomas and gastrinomas.
Insulinomas are tumours of insulin-producing beta cells of the pancreas. The excessive insulin release leads to a hyperinsulinaemic state and patients may present with episodes of severe hypoglycaemia. It should be suspected in the presence of high serum insulin and hypoglycaemia.
Gastrinomas are tumours of gastrin secreting G cells, which leads to excessive gastric acid secretions due to stimulation of parietal cells. Zollinger-Ellison syndrome is a condition characterised by a gastrinoma and recurrent peptic ulcers commonly associated with diarrhoea. Symptoms are mainly due to the peptic ulcers whilst the diagnosis is based on high serum gastrin levels with ulceration seen on endoscopy.
Diagnosis
MEN 1 is defined as the presence of two or more MEN 1 tumour types.
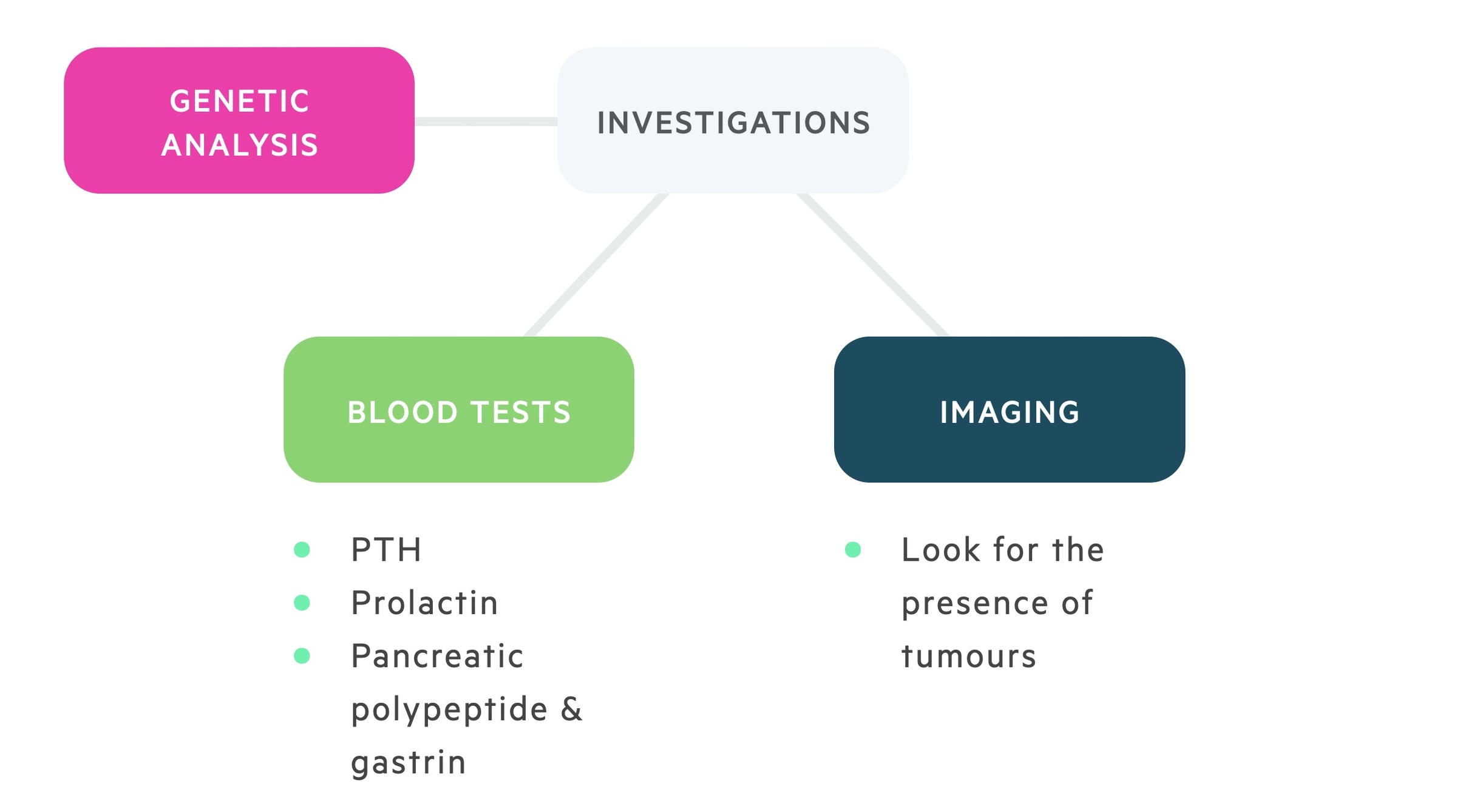
Those with a known family history (first and second-degree relatives) may be screened. Screening tends to be annual, typically from the ages of 5 to 65 (and may continue after this time, but less frequently). There is a significant amount of variability in screening protocols.
Hormonal tests
Serum calcium (to detect asymptomatic hypercalcaemia) is the test of choice due to the high incidence of primary hyperparathyroidism in those with MEN 1. Other hormonal tests include parathyroid hormone, prolactin and gastrin levels.
Imaging
Imaging with MRI and CT may be useful in the screening and diagnosis of MEN 1. Again there is no single protocol that all clinicians follow. There is no clear evidence that diagnosis of these tumours prior to clinical or biochemical detection offers any survival benefit.
The risks of repeated exposure to radiation (in the case of CT scanning) and the cost of these programmes should be considered.
DNA testing
Unlike MEN 2 there is no clear evidence of the benefit of using DNA testing in the general screening process of MEN 1.
The sensitivity of DNA testing is highly variable, it is detectable in far less sporadic cases than familial ones. There are settings where it is indicated such as the preimplantation of an embryo. DNA testing may also be utilised in those presenting with a characteristic tumour.
Management
Management is dependent on the tumour type and symptomatic status.
Primary hyperparathyroidism
Primary hyperparathyroidism is managed surgically with a parathyroidectomy. Surgery is indicated in those with pathological features of excess PTH and a raised serum calcium. Such features include osteopenia, nephrolithiasis and symptomatic hypercalcaemia.
There is a high incidence of recurrence following subtotal parathyroidectomy. More ‘aggressive’ initial operations reduce this risk but increase the development of hypoparathyroidism.
Many advocate cervical accessible thymectomy, a partial thymectomy, beneficial as the thymus often contains parathyroid tissue.
Pituitary adenomas
The treatment of pituitary adenomas is dependent on the cell line affected and whether it is a macro or microadenoma.
Management is the same as for sporadic cases. Both medical and surgical options are available. When surgery is indicated this typically takes the form of transsphenoidal resection.
Pancreatic tumours
Pancreatic neuroendocrine tumours are the main cause of morbidity and mortality in MEN 1.
Insulinomas may be treated pharmacologically with diazoxide (inhibits insulin release). Surgery is the definitive management. It is complicated by the fact that multiple insulinomas may be present requiring a subtotal pancreatectomy.
Gastrinomas may be treated pharmacologically with proton pump inhibitors. Surgical excision is indicated in those who do not maintain symptomatic relief, with large tumours or wish for surgical management.
MEN type 2
MEN 2 is an autosomal dominant condition with a frequency of 1 in 30,000.
It is caused by a mutation of the RET proto-oncogene. The condition is predominantly characterised by primary hyperparathyroidism, medullary thyroid cancer and phaeochromocytoma.
- MEN 2A: normal appearance, primary hyperparathyroidism is common.
- MEN 2B: marfanoid habitus, primary hyperparathyroidism is rare.
1. Primary hyperparathyroidism
Primary hyperparathyroidism occurs in around 10-25% of patients with MEN 2. It is seen in type 2A but is not associated with type 2B.
Parathyroid hormone has multiple effects on calcium homeostasis including activation of vitamin D, increasing serum calcium levels. Patients may present with symptoms of hypercalcaemia but in MEN 2 it is frequently mild and asymptomatic.
For more information, see our notes on Primary hyperparathyroidism.
2. Phaeochromocytomas
Phaeochromocytomas are neuroendocrine tumours arising from the adrenal medulla. These tumours cause an excess of the catecholamines adrenaline and noradrenaline.
In MEN 2 phaeochromocytomas are bilateral in around 60% of cases as opposed to 10% of sporadic cases. They are rarely malignant and tend to occur in the second to third decades of life.
For more information, see our notes on Phaeochromocytoma.
3. Medullary thyroid cancer
Medullary thyroid cancer occurs in almost all patients with MEN 2. The disease is characteristically bilateral and multifocal in MEN 2, as opposed to unilateral disease seen in sporadic cases.
Due to high penetrance, prophylactic surgery is indicated in all patients with MEN 2.
For more information, see our notes on Medullary thyroid cancer (though this note predominantly focuses on papillary and follicular thyroid cancer).
Diagnosis
MEN 2 should be considered in those presenting with a characteristic tumour, particularly if young with multifocal disease.
MEN 2A is defined by the identification of the RET oncogene mutation or the presence of two or more of the characteristic tumour types. Genetic testing is always indicated in index cases.
Those with MEN 2 and their family members require screening for the development of tumours. Screening allows for early diagnosis and treatment reducing morbidity and mortality.
Those who present with MTC should be screened for phaeochromocytomas with plasma fractionated metanephrines and hyperparathyroidism with serum calcium.
Those not presenting with MTC should have serum calcitonin levels measured and an ultrasound of the neck.
Management
Prophylactic thyroidectomy is indicated in patients with confirmed MEN 2.
Primary hyperparathyroidism
Primary hyperparathyroidism is managed surgically with a parathyroidectomy. Surgery is indicated in those with pathological features of excess PTH and a raised serum calcium. Such features include osteopenia, nephrolithiasis and symptomatic hypercalcaemia.
The risk of recurrence is lower than that seen in MEN 1.
Phaeochromocytomas
Phaeochromocytomas must be managed surgically with either unilateral or bilateral adrenalectomy.
Those receiving a bilateral adrenalectomy require glucocorticoid and mineralocorticoid replacement to manage the resulting adrenal insufficiency.
Medullary thyroid cancer
Patients with an identified RET oncogene mutation (and yet to develop MTC) should receive a prophylactic thyroidectomy.
Those diagnosed with MTC should receive a thyroidectomy and lymph node dissection as appropriate.
Calcitonin and carcinoembryonic antigen should be used to monitor for recurrence.

