Introduction
Henoch-Schönlein purpura is an IgA vasculitis, which commonly presents in childhood.
Henoch-Schönlein purpura (HSP) is the most common vasculitis of childhood. Approximately, 90% of cases occur in children. The condition commonly presents in children aged 3-15 years old with an annual incidence of 20 per 100,000 in children aged < 17 years old.
HSP is usually precipitated by a viral illness, leading to an immune-mediated reaction in blood vessels. The majority of cases are self-limiting without longterm sequelae. Similar to other vasculitidies, it is characterised by the presence of purpura. This refers to small haemorrhages within the skin approximately 3-10 mm in size.
Aetiology
HSP is an immune-mediated disorder due to deposition of the IgA immunoglobulin in blood vessels.
The exact cause for the deposition of IgA and subsequent development of HSP is unknown. It is estimated that up to 50% of children have a preceding upper respiratory tract infection, particularly following Streptococcal infections.
The association with infections suggests the autoreactive IgA antibodies (i.e. antibodies that attack our bodies own antigens), may have originally been created as part of the normal immune response.
Pathophysiology
Vasculitis refers to inflammation of blood vessels.
HSP is characterised by a leukocytoclastic vasculitis (LCV). LCV is generally a histopathological term that refers to a small-vessel vasculitis, but may be used to refer to cutaneous vasculitis. LCV classically presents with palpable purpura within the skin. The vasculitis may be localised to the skin or associated with other organ involvement (e.g. kidneys, GI tract, joints).
HSP is one particular cause of LCV due to deposition of IgA immune complexes. This refers to deposition of antibody-antigen complexes within the basement membrane of blood vessels that can activate complement and other inflammatory pathways. Other precipitants of LCV include medications, infections, collagen vascular disease (e.g. rheumatoid arthritis) and malignancy.
Clinical features
Palpable purpura in a symmetrical distribution over the lower limbs is typical of HSP.
The characteristic purpuric skin rash is the presenting feature in up to 75% of cases. It may be pruritic, but rarely painful. It usually appears as crops in a symmetrical distribution over pressure-dependent areas and extensor surfaces of the lower limbs (e.g. buttocks, knees). May appear as small petechiae (1-2 mm), purpura (3-10 mm) or coalesce to form ecchymosis (>10 mm).
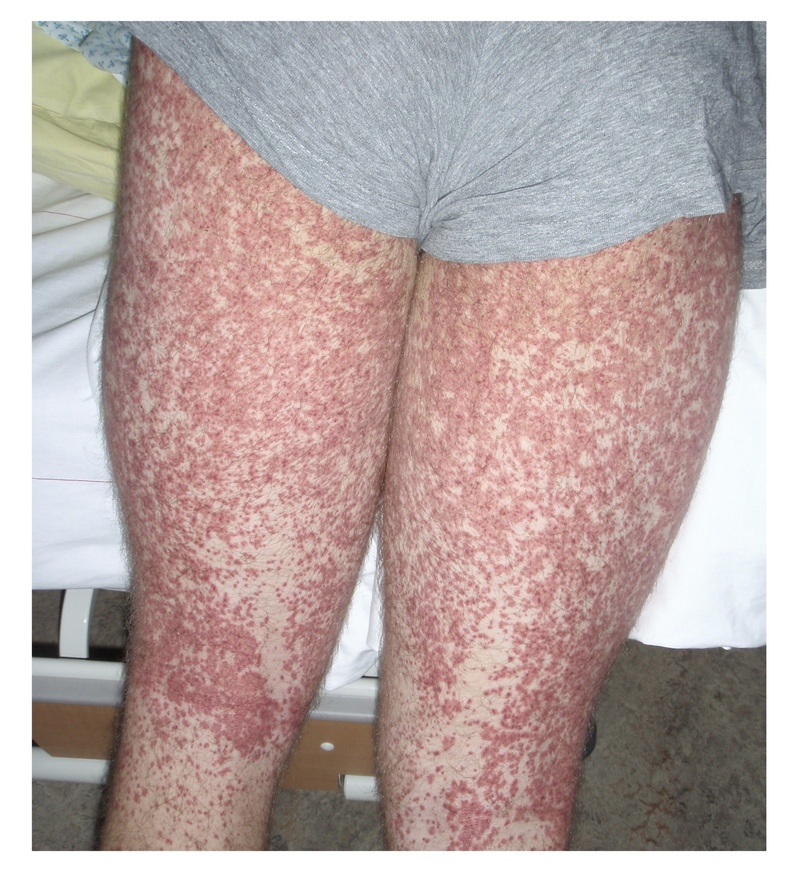
Typical purpuric rash of HSP on lower limbs.
Image courtesy of Peter Rammstein (Wikimedia commons)
Possible clinical presentations
- Arthritis/arthralgia (>80%): migratory and small number affected (1-4).
- Gastrointestinal (GI) upset (>50%): nausea, vomiting and pain. Usually 8 days following rash.
- Severe GI features: GI bleeding, intussusception, severe pain from perforation/peritonitis.
- Haematuria with mild/absent proteinuria: more common in older children/adults.
- Nephritic syndrome: oliguria, haematuria and hypertension with renal impairment.
- Other: headache, seizures, encephalopathy, testicular pain/orchitis, coma.
Diagnostic criteria
The diagnosis of HSP is based on the EULAR and PRES diagnostic criteria.
The diagnostic criteria for HSP was originally developed by both the European League Against Rheumatism (EULAR) and the Paediatric Rheumatology European Society (PRES). It is based on the presence of palpable purpura, which is a mandatory finding, and at least one other clinical criteria.
- Mandatory: palpable purpura in absence of thrombocytopenia or coagulopathy.
- Non-mandatory (must have at least one): diffuse abdominal pain, acute arthritis/arthralgia, renal involvement (haematuria/proteinuria), biopsy showing LCV or proliferative glomerulonephritis with predominant IgA deposition
Differential diagnosis
Always consider other causes of a non-blanching purpuric/petechial rash.
Key differentials for a non-blanching rash including sepsis (e.g. meningococcal bacteraemia) and immune thrombocytopaenic purpura (ITP). ITP is an autoimmune condition due to immune-mediated destruction of platelets. It is usually self-limiting in children following a viral illness, whereas in adults it may require immunosuppressive therapy. The key difference with HSP is the abnormal platelet count.
Other causes of a non-blanching rash include medication-induced thrombocytopaenia, bone marrow failure (e.g. due to haematological malignancies or aplastic anaemia), connective tissues disease due to vasculitis and coagulopathies (e.g. disseminated intravascular coagulation).
Investigations
The diagnosis is usually suspected clinically, but investigations help rule out other pathologies and look for organ involvement.
Bedside
- Urine dipstick: blood/protein
- Protein/creatinine ratio (PCR)
- Blood pressure
Bloods
- FBC: essential to check haemoglobin and platelet count
- Blood film: look for other causes of purpura (i.e. malignancy)
- Coagulation
- If infection concern: CRP and blood cultures
- Routine: U&Es, Bone profile, LFTs
- ASOT*
- Autoimmune screen (if diagnosis in doubt): complement, immunoglobulins, ANA, ANCA, dsDNA
Imaging
- Not routinely required: consider abdominal imaging (e.g. ultrasound) if diagnosis in doubt or severe GI symptoms
Special
- Skin/renal biopsy: reserved for atypical presentations in paediatric population
*NOTE: ASOT refers to anti-streptolysin O titres. May be suggestive of current or recent Streptococcal infection
Management
The majority of children with HSP will make a full recovery and can be monitored as an outpatient.
Most cases of HSP are self-limiting over a period of 6 weeks. Children will only require supportive therapy, which includes good oral intake, rest and analgesia.
Paracetamol and NSAIDs can be prescribed for pain control of joint and abdominal pain. This usually resolves quickly. However, if pain is severe, it is important to consider complications (e.g. intussusception) and steroids may be given with senior input. Steroids may shorten duration of symptoms but do not effect the clinical course.
Some children may require admission due to complications or inability to maintain oral intake.
Admission criteria
- Hydration: unable to maintain adequate hydration with oral intake
- Pain: severe abdominal pain or joint pain limiting mobility/self-care
- GI complications: bleeding
- Altered mental status
- Renal disease: AKI, hypertension, and/or nephritis/nephrotic syndrome
Specialist input
Some children require surgical input if they development abdominal complications (e.g. GI bleeding, perforation, intussusception). Others need input from a paediatric nephrologist in cases of nephritis/nephrotic syndrome, impaired renal function, or hypertension.
Prognosis
Approximately, 1% of children will progress to end-stage renal disease.
Renal involvement will develop within the first 6 months in up to 97% of patients.
Collectively, two-thirds of children have a self-limiting illness without relapse. One third of children will relapse, particularly those who initially presented with renal disease. A single recurrence is most common, but some children can have multiple relapses. Ongoing follow-up in primary or secondary care may be required according to local guidance.

