Overview
Biliary atresia refers to progressive destruction of the extrahepatic biliary system.
Biliary atresia presents exclusively in the neonatal period with biliary obstruction. It is due to progressive destruction of the extrahepatic biliary system. The cause is idiopathic and may be associated with other congenital malformations.
It is the most common reason for surgical intervention in neonatal jaundice and the most common cause of liver transplantation in children. The incidence is around 1 in 10,000-20,000 live births.
Without surgical intervention to fix the obstruction, the condition is invariably fatal.
Aetiology and pathophysiology
The cause of biliary atresia is idiopathic.
Biliary atresia is a phenotype that can occurs due to a variety of injuries to the biliary system of the fetus during development or the perinatal period. The cause is idiopathic, but there are some suspected underlying aetiological factors including viral, genetic, toxic and immune-mediated.
- Viral (infectious): temporal association with viral infections. Specific virus has not been implicated
- Genetic: certain types of biliary atresia linked to genetic defects (e.g. CFC1 variant in biliary atresia and splenic malformations).
- Toxic: environmental toxics have been implicated in biliary atresia in animal models.
- Immune: a primary immune disorder or a dysregulated immune response to a infectious or genetic trigger has been implicated.
Types of biliary atresia
Biliary atresia can be further classified based on biliary anatomy, clinical presentation or association with other abnormalities.
The Ohi classification divides the types of biliary atresia into three groups based on anatomy. These can be further subdivided based on proximal and distal anatomical features of the biliary tree.
- Type 1: common bile duct atresia
- Type 2: atresia of hepatic ducts
- Type 3: porta hepatis atresia (entry point of vessels and ducts to liver). Most common.
Several congenital malformations may associate with biliary atresia including situs inversus, asplenia, intestinal atresia, or imperforate anus to name a few. Newborns with other congenital malformations are more likely to have an embryonic form of biliary atresia and present with jaundice at birth.
Clinical features
Biliary atresia presents with jaundice, which can occur any time from birth to 8 weeks.
Biliary atresia is a classic ‘surgical’ cause of neonatal jaundice. It is unlikely to appear after 8 weeks.
Features
- Jaundice (yellow pigmentation of skin and mucous membranes)
- Pale stools
- Dark urine
- Hepatomegaly (may be present)
- Associated congenital malformations
Presentations
- Embryonic form: jaundice at birth, usually associated with other congenital abnormalities.
- Perinatal form: jaundice can develop any time following birth. Unlikely after 8 weeks.
Diagnosis & investigations
A definitive diagnosis of biliary atresia is made by an intraoperative cholangiogram showing biliary obstruction.
Biliary atresia is a cause of prolonged jaundice, which is jaundice occurring for >14 days. The key investigation is split bilirubin, which looks at both the conjugated and unconjugated components. Conjugated bilirubin (i.e. direct bilirubin) is a marker of biliary obstruction, which is seen in biliary atresia.
Investigations confirm conjugated hyperbilirubinaemia and screen for other causes:
- Split bilirubin: conjugated bilirubin >17.1 umol/L abnormal
- Metabolic screen (inc. TFT, cystic fibrosis): look for inborn errors of metabolism
- FBC
- LFTs
- Others (depending on results and suspicion)
If biliary atresia is suspected a series of additional investigations are needed to confirm the diagnosis:
- Abdominal ultrasound: typically non-diagnostic. Helps exclude external compression (e.g. cyst)
- Hepatobiliary scintigraphy: nuclear medicine scan looking for excretion of a radioactive tracer into the bowel via the biliary system. Helps to exclude biliary atresia if normal excretion. However, absent excretion does not confirm the diagnosis.
- Liver biopsy: helps look at characteristic changes of biliary atresia and exclude other causes.
- Cholangiogram: passage of contrast through the biliary system. Confirms obstruction. Gold standard.
NOTE: a cholangiogram is usually performed intraoperatively. It guides the decision for surgical management.
Management
The initial management of biliary atresia is a hepatoportoenterostomy, known as the Kasai procedure.
The Kasai procedure aims to restore bile flow from the biliary system to the intestines. A roux-en-Y loop of small intestine is anastomosed directly to the hilum of the liver after excision of the remnant biliary system. The proximal end of the intestine at the point of excision is then rejoined side-on.
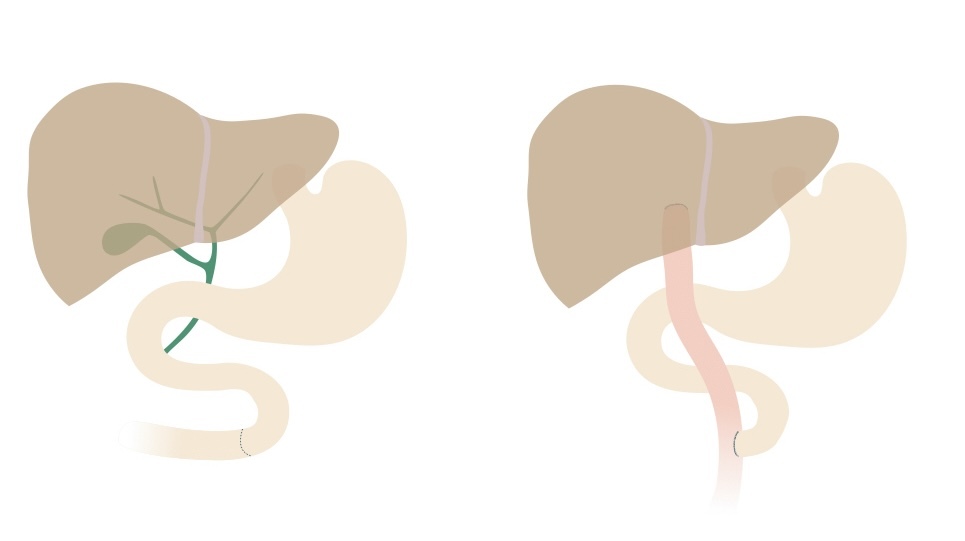
Kasai procedure
NOTE: a roux-en-Y loop is a typical general surgical procedure. It involves cutting a point in the small bowel. The distal end is dragged up and connected to a proximal organ (e.g. stomach, liver). The proximal end is rejoined to the intestine side-on.
A Kasai procedure should be performed within 45-60 days of life. The majority of patients will ultimately require a liver transplantation due to slowly progressive liver disease. At least 50% will need a liver transplant by two years old.
Prognosis
The combination of Kasai procedure and liver transplantation means >90% survive into adulthood.
It is estimated that 60-80% of patients with biliary atresia will ultimately need a liver transplantation. The main indications for transplant include portal hypertension, progressive liver dysfunction and/or growth failure.
The five-year survival for liver transplantation after two years old is estimated at 97%.

