Overview
Multiple sclerosis is a chronic, immune-mediated inflammatory disease of the central nervous system.
Multiple sclerosis (MS) is a demyelinating neuroinflammatory condition, which affects the central nervous system (CNS). The CNS comprises the brain, brainstem and spinal cord. The condition is most commonly seen in young adults.
Demyelination, which is the hallmark of MS, refers to damage to the protective myelin sheath that surrounds neurons. This can lead to scarring and secondary neuronal cell loss, which results in irreversible neurological damage.
Due to the wide area of the CNS that can be affected, MS can present with a range of clinical manifestations and the disease course between individuals is unpredictable. MS is most commonly characterised by a relapsing-remitting course, which may lead to progressive neurological disability overtime.

Epidemiology
MS is 2-3 times more common in women than men.
MS is predominantly a disease of young adults with a mean age of onset around 30 years old. It is estimated that 70% of patients with MS will present between 20-40 years old.
In the UK prevalence varies widely depending on location from a low of 179 per 100,000 in Wales to a high of 290 per 100,000 in Scotland.
Aetiology & risk factors
The aetiology of MS remains poorly understood.
Like most inflammatory conditions of unknown aetiology, the cause is thought to be due to an abnormal immune reaction to an unknown environmental trigger in a genetically predisposed individual.
Genetics
There is evidence of genetic susceptibility in MS with more than 200 polymorphisms (i.e. genetic variations).
In twin studies, the concordance rate is around 20-35% in monozygotic twins. Concordance means if one twin develops MS, what is the likelihood that the other twin, who has the same genetic material, also develops the condition. At 20-35%, it suggests genetics only play a modest role in the development of MS. The concordance rate in dizygotic twins of only 5%.
Risk factors
When the aetiology of a condition is unknown, we look at different risk factors that may be associated with triggering an autoimmune response that subsequently leads to the disease.
Several risk factors have been linked to the development of MS including genetics (as discussed), viral infections, geographic latitude, sunlight exposure and vitamin D levels among others.
- Viral infections: several viruses linked with MS. Increased risk of MS following Epstein-Barr virus (EBV) infection (i.e. glandular fever)
- Geographic latitude: prevalence of MS increases the greater the distance north or south from the equator. Migration after puberty carries risk from former geographic location.
- Sunlight exposure: inverse relationship between MS, sunlight exposure and vitamin D levels.
- Other: obesity during adolescence, smoking, gender (females at increased risk).
Pathophysiology
MS is an inflammatory, demyelinating disease of the CNS, which is characterised by the presence of plaques.
The CNS is comprised of neurons, which contain a cell body and axons. Axons extend out and make connections with other neurons at synaptic junctions. These neurons are supported by glial cells, which are the most abundant cell type in the CNS.
One group of glial cells, known as oligodendrocytes, are important in formation of the myelin sheath that surrounds and insulates neuronal axons. In MS, oligodendrocytes are destroyed, which leads to demyelination and can cause axonal loss. Although there is no direct proof for an autoimmune cause, there is mounting evidence for involvement of the immune system in this pathological process.
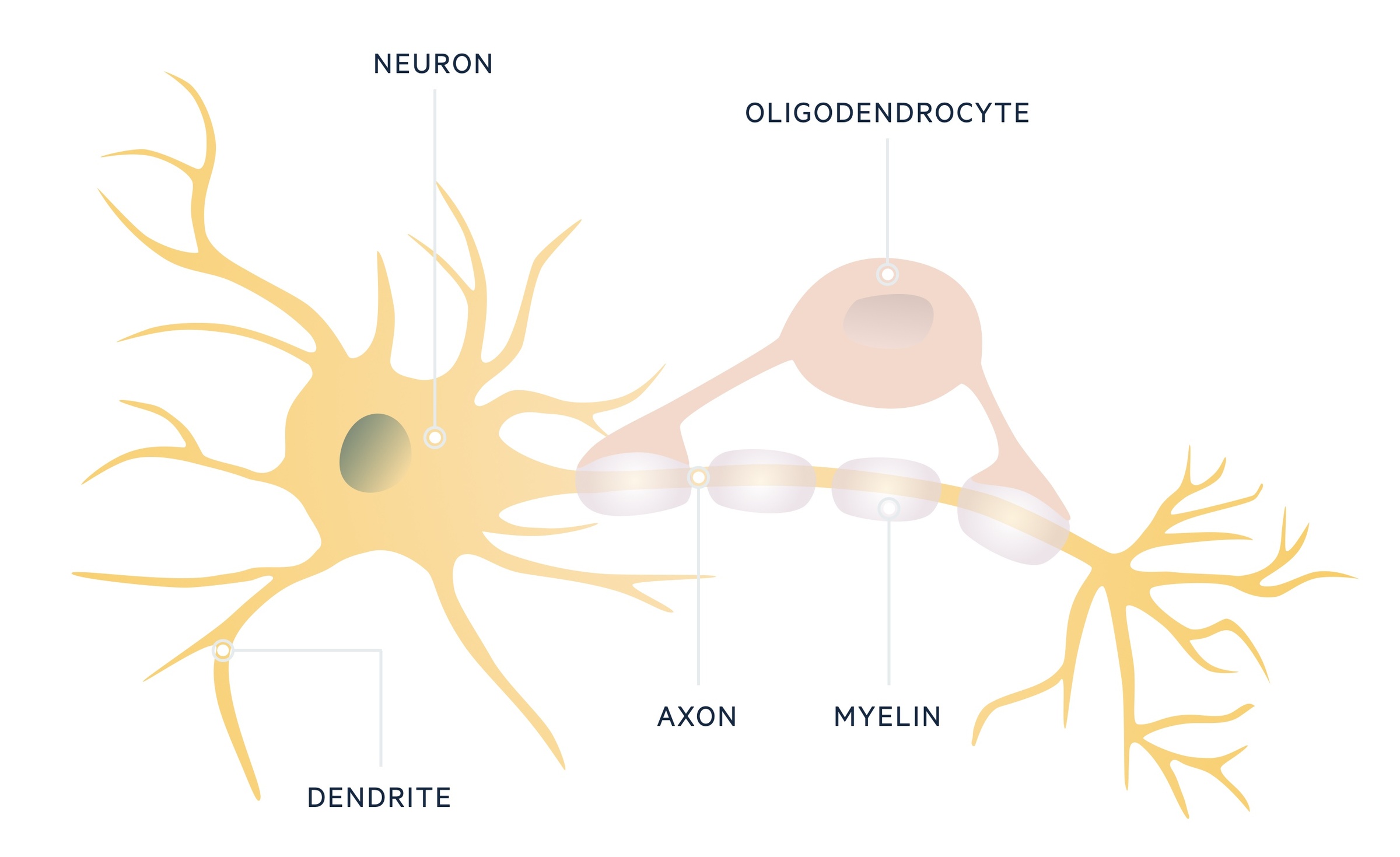
Pathological process
The exact mechanism of MS is still to be elucidated. However, there appears to be activation of myelin-reactive T lymphocytes and disruption of the blood brain barrier (BBB), which allows entry of autoreactive immune cells.
Within the CNS, there is then a pro-inflammatory response with recruitment of further inflammatory cells including B lymphocytes, macrophages and microglia. Microglia can be considered as ‘macrophages of the CNS’ and thus critical in cytokine release, phagocytosis and antigen-presentation.
There is a marked immune response including an antibody-mediated response with evidence of immunoglobulins, termed oligoclonal bands, in the cerebrospinal fluid (CSF). The continued immune reaction leads to damage to oligodendrocytes with subsequent demyelination and formation of ‘MS plaques’, which contain myelin reactive T cells, B cells and macrophages.
Within the focal areas of demyelination, or plaques, there is variable degrees of inflammation, scarring (i.e. gliosis) and axonal injury. The clinical manifestations of MS depend on the location of these plaques within the CNS.
Classical plaque sites
- Optic nerves: affects 40% during course of disease. Presenting demyelinating event in 20%.
- Spinal cord: affects 50-75% during course of disease. Majority associated with concomitant brain lesions. Predilection for cervical spine.
- Brainstem: may present with ophthalmoplegia (e.g. intranuclear ophthalmoplegia – discussed below).
- Cerebellum: characteristically causes ataxia and gait disturbance.
- Juxtacortical white matter (near the cerebral cortex).
- Periventricular white matter (near the ventricular system).
Classification
Up to 90% of cases of MS are characterised by a relapsing-remitting disease course.
MS can be categorised into different types depending on the disease course:
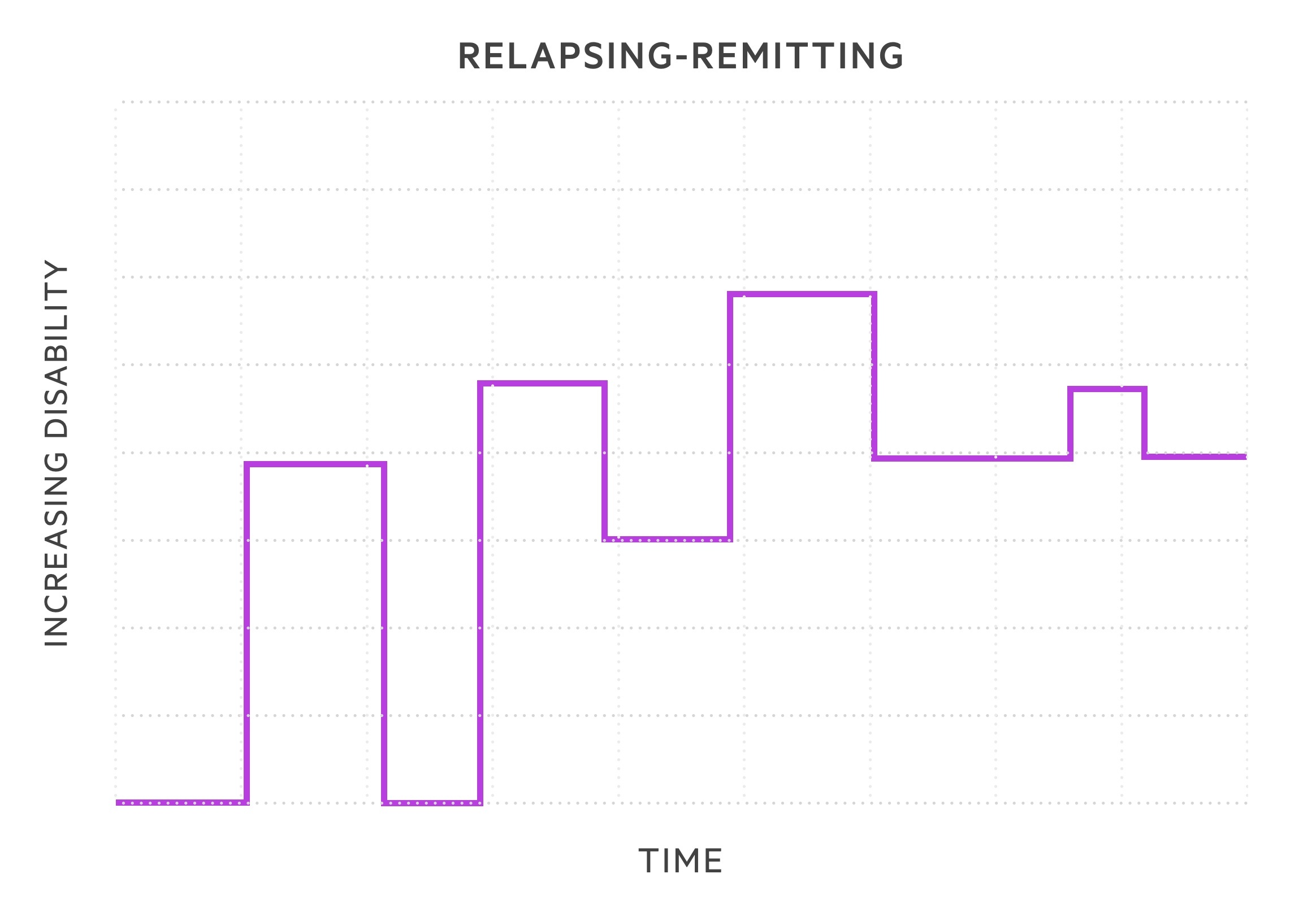
- Relapsing-remitting MS (RRMS): subtype in 85-90% of cases. Episodes of exacerbation in symptoms termed relapses followed by periods of recovery termed remissions. During early stages, symptoms may completely remit. As diseases progresses likely to retain residual damage with each relapse.
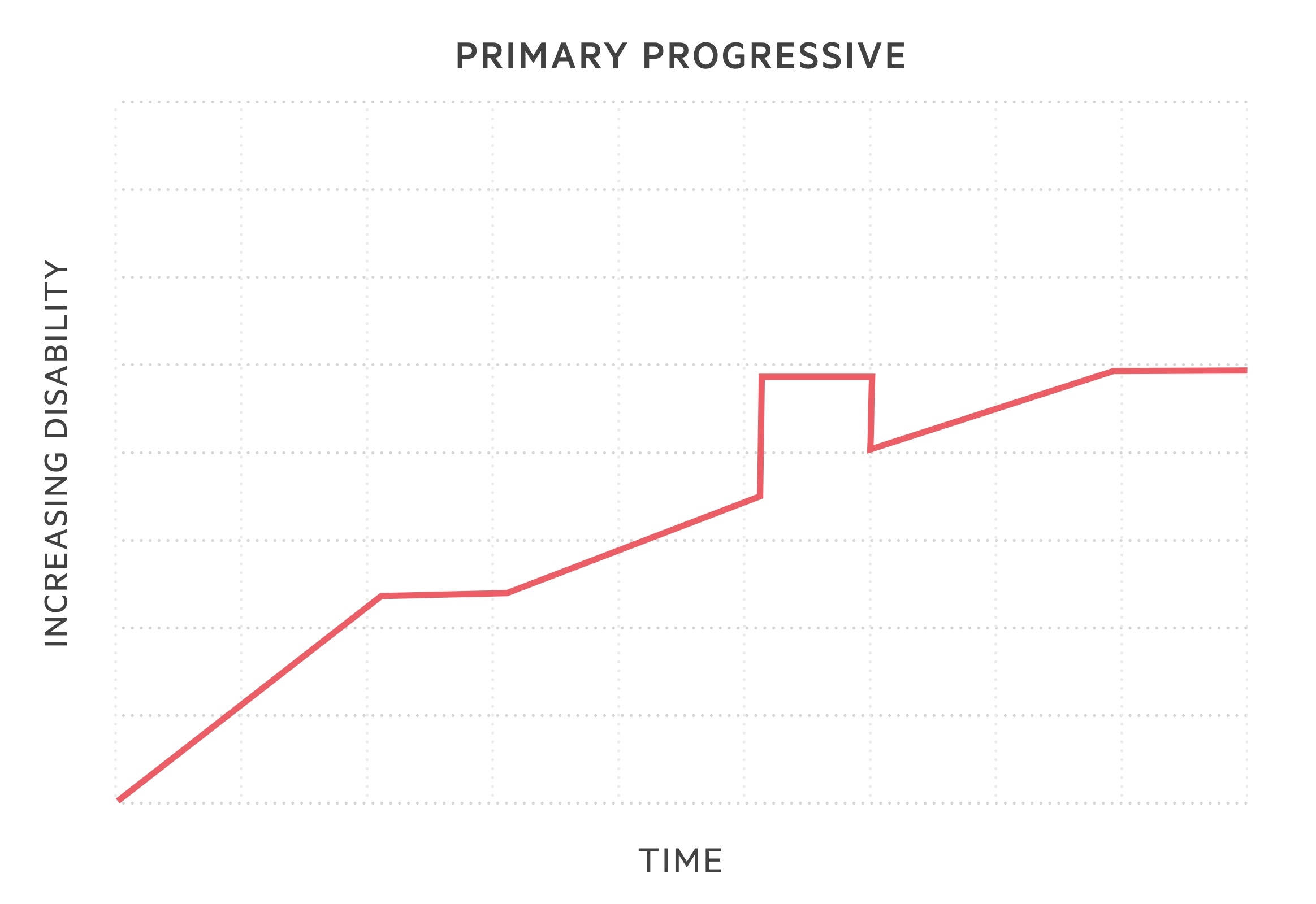
- Primary progressive MS (PPMS): subtype in 10-15%. Sustained progression of disease severity from onset. May have periods where disease is not active or non-progressive, but no evidence of clinical remission.
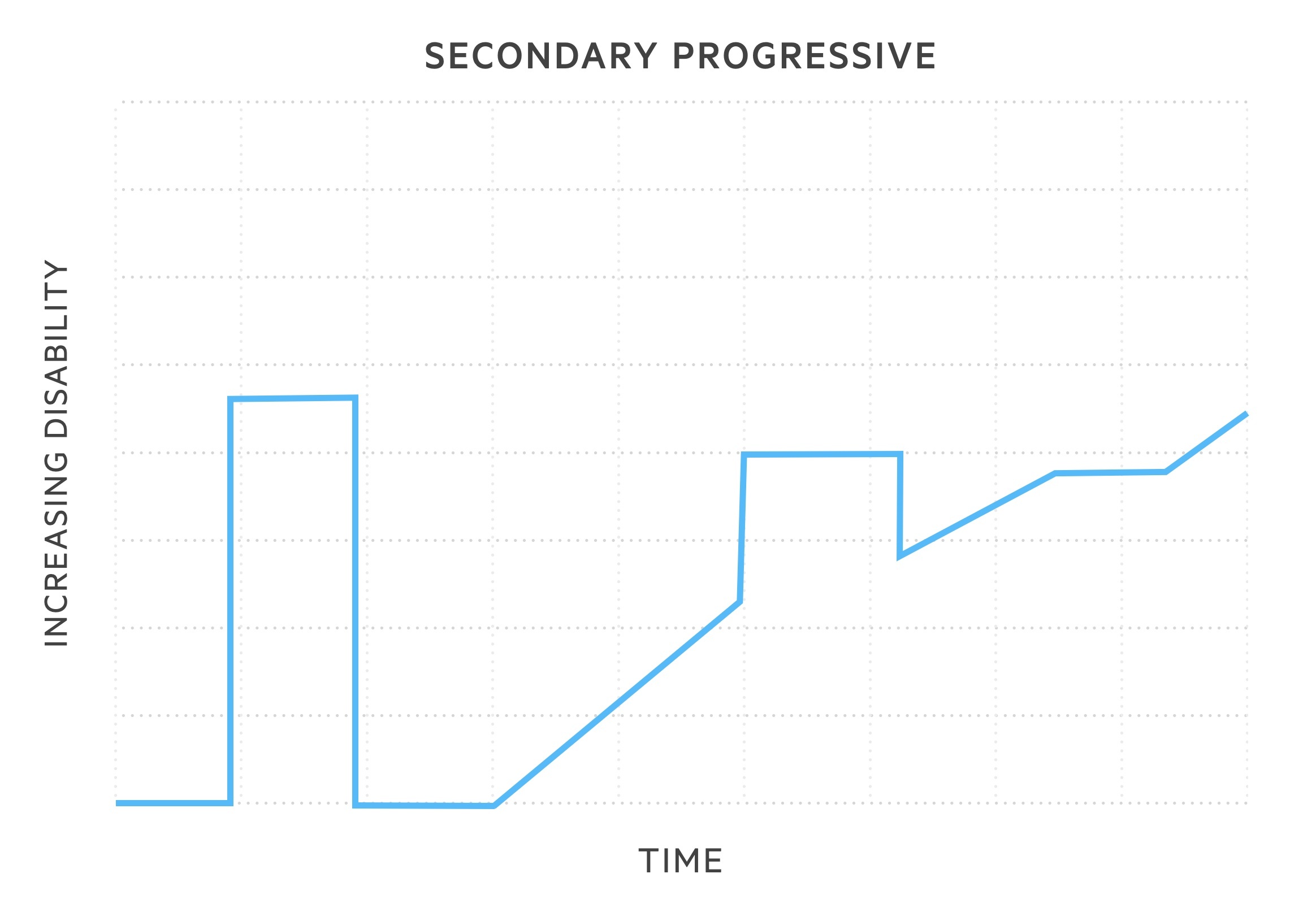
- Secondary progressive MS (SPMS): 50% of patients with RRMS will develop this subtype within 15 years of onset. Following RRMS phenotype, disease course changes with gradual, sustained worsening in neurological function. Relapses may still occur but without remission.
Clinically isolated syndrome (CIS)
CIS describes the first clinical episode of suspected MS. There is no previous evidence of demyelination clinically or on neuroimaging. Oligoclonal bands in cerebrospinal fluid may be used as supportive criteria to help come to the diagnosis (see supporting investigations).
Clinical manifestations
Clinical features associated with MS are determined by plaque locations within the CNS.
There are a number of clinical features that are characteristic of MS including optic neuritis, Lhermitte phenomenon (uncomfortable electric shock-like feeling triggered by neck flexion) and internuclear ophthalmoplegia (INO). Others may be common to numerous neurological pathologies including motor weakness, diplopia, gait disturbance and bladder/bowel dysfunction.
We can broadly divide the clinical manifestations into four groups: visual, motor & coordination, sensory & autonomic and cognitive & psychological.
Visual
Optic neuritis and eye movement abnormalities commonly affect patents with MS.
Optic neuritis is due to inflammation of the optic nerve and characteristically presents with partial or total unilateral visual loss that develops over days. Typical features include:
- Visual loss
- Blurred vision
- Pain: typically behind the eye and on movement
- Scotoma: refers to partial visual field loss
- Poor colour differentiation
- Relative afferent pupillary defect
- Optic nerve swelling: seen on fundoscopy
Eye movement disorders may be due to brainstem lesions that affect cranial nerves or the pathways that connect visual tracts together. Two commonly seen are INO and Abducens palsy.
- INO: disorder of conjugate lateral gaze due to demyelination of the medial longitudinal fasciculus (MLF). MLF is heavily myelinated. Connects the abducens nucleus complex with the contralateral oculomotor nucleus. If affected, when looking to the right, the right eye will abduct but the left will remain central (failure in adduction).
- Abducens palsy: absence of lateral abduction of the eye. If the right abducens nerve is affected, when looking to the right, the right eye will remain central.
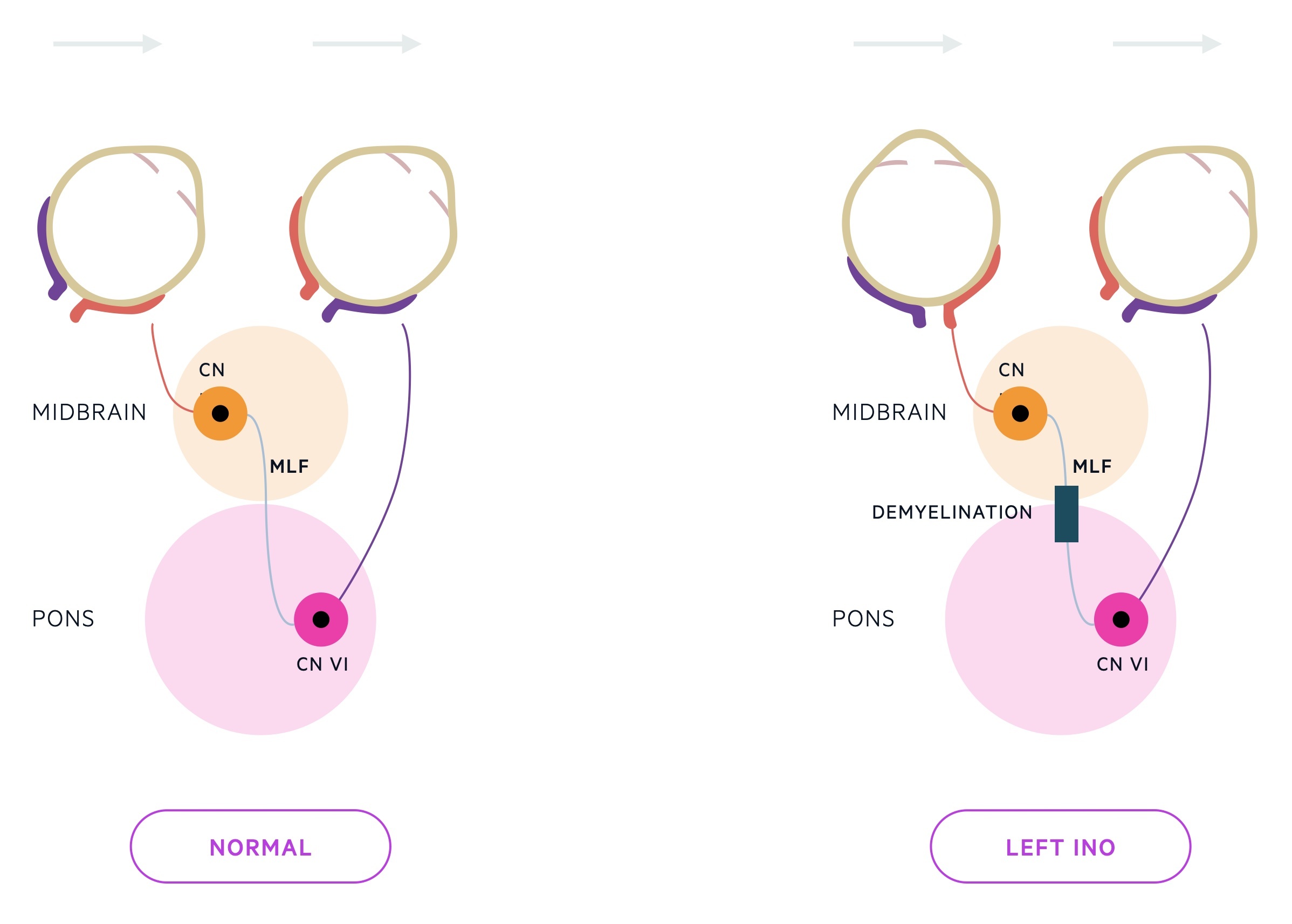
Motor & coordination
Motor abnormalities can occur in any part of the body, but usually characterised by weakness and ataxia (lack of muscle coordination). The prominent motor feature in MS is progressive paraparesis and evidence of upper motor neuron signs (Spasticity, reduced power, hyper-reflexia).
Motor and coordination abnormalities may present as a typical clinical syndrome such as transverse myelitis or cerebellar syndrome.
- Transverse myelitis: focal inflammation within the spine. Characterised by sensory and motor symptoms below the level of the lesion (e.g. paraesthesia, weakness). Bladder/bowel involvement may be present and usually there is a sensory level that corresponds to the level of the lesion. Must exclude compressive pathology in first instance (i.e. metastatic cord compression).
- Cerebellar syndrome: a clinical syndrome characterised by disease of the cerebellum, which manifests with ataxia, slurred speech, intension tremor, nystagmus, vertigo and clumsiness.
Sensory & autonomic
Sensory symptoms are common in MS and seen in almost all patients during the course of the disease.
There are a whole range of sensory and autonomic symptoms that should be explored, including:
- Paraesthesia
- Pain
- Heat sensitivity (Uhthoff phenomenon)
- Sexual dysfunction
- Bladder & bowel dysfunction
NOTE: Up to 75% report bladder dysfunction. A range of Neurogenic bladder dysfunction may occur leading to incontinence, poor bladder emptying or urinary retention.
Cognitive & psychological
Cognitive impairment, depression and fatigue are all extremely common in MS. It is estimated that some degree of depression may affect up to two thirds of patients with MS and this can have a negative impact on memory, attention, and concentration.
Diagnosis
MS is primarily a clinical diagnosis, which is supported by the use of MRI.
Patients with suspected MS should be referred to a neurologist who specialises in neuroinflammatory disorders. A series of investigations can be arranged prior to referral to help exclude an alternative diagnosis (see section supporting investigations).
The diagnosis of MS is based on the clinical presentation (called an ‘attack’ or ‘relapse’), which is then confirmed by objective clinical evidence.
- MS attack: defined simply as an episode of neurological symptoms that relate to an inflammatory demyelinating lesion. Lasts > 24 hours with or without recovery. Typically sensory disturbances, motor weakness, or visual complaints. There must be more than 30 days between attacks to count as a separate episode.
- Objective clinical evidence: identification of an abnormality (usually on clinical examination or MRI), which corresponds to the anatomical location suggested by the current, or past, symptoms of a MS attack.
Revised McDonald criteria
The McDonald criteria provides recommendations for the diagnosis of MS based on MRI findings (brain +/- spinal cord) and clinical presentation. MRI is the imaging of choice for identification of demyelinating lesions within the CNS.
The McDonald criteria was revised in 2017 and it is based on the principle that demyelinating lesions are disseminated in time and space. In other words, two independent clinical attacks would be evidence of dissemination in time and two separate lesions on MRI would be evidence of dissemination in space.
The criteria can get quite niche, but these general principles apply:
- ≥2 attacks with objective clinical evidence of ≥2 lesions: MS diagnosed
- ≥2 attacks but objective clinical evidence for only one lesion: evidence of dissemination in time, but not space. MS diagnosed if dissemination in space shown on MRI or subsequent clinical attack representing new area.
- Single attack with objective clinical evidence of one lesion: clinically isolated syndrome. MS diagnosed once proof of both dissemination in time and space.
- Single attack with objective clinical evidence of ≥2 lesions: clinically isolated syndrome. MS diagnosed once proof of dissemination in time.
NOTE: MRI may be used to show dissemination in time by the simultaneous presence of gadolinium-enhancing and nonenhancing lesions at any time, or by a new gadolinium-enhancing lesion(s) on follow-up MRI
Differential diagnosis
The differential diagnosis of MS depends on the site demyelination and presentation of neurological symptoms.
There are numerous conditions that present in a similar manner to MS. These may be other demyelinating conditions, infections, malignancy, metabolic disorder or even systemic inflammatory conditions.
- Demyelinating diseases:
- Neuromyelitis optica
- Transverse myelitis
- Acute disseminated encephalomyelitis
- Infections:
- Lyme disease
- Human T-lymphotropic virus
- Tertiary syphilis
- Metabolic diseases:
- B12 deficiency
- Diabetic neuropathy
- Adult-onset leukodystrophies
- Copper deficiency
- Systemic diseases:
- SLE
- Sarcoidosis
- Other:
- Neoplasia
- Vasculitis
- Stroke
Neuromyelitis optica (Devic’s disease)
Neuromyelitis optica spectrum disorders (NMOSD) is the new nomenclature for neuromyelitis optica, a condition with a very similar phenotype to MS. It refers to a series of neuroinflammatory demyelinating conditions, which preferentially affect the optic nerve and spinal cord.
The hallmark of NMOSD is an acute attack of bilateral optic neuritis or transverse myelitis. These are two common clinical manifestations of MS, but in NMOSD the presentation is more severe. It is associated with formation of an IgG autoantibody to aquaporin-4 (AQP4). Treatment is similar to MS with corticosteroids, but plasma exchange may be required for refractory cases.
Supporting investigations
Additional investigations can be used to help support the diagnosis and exclude an alternative pathology.
Bloods
- Full blood count
- CRP/ESR
- LFTs, U&Es, Bone profile
- Glucose, HbA1c
- TFTs
- Haematinics
- Blood-borne virus screen
Oligoclonal bands
Cerebrospinal fluid (CSF) can be analysed following a lumbar puncture to assess for CSF-specific oligoclonal bands.
Oligoclonal bands refer to bands of immunoglobulins. The test is considered positive if oligoclonal bands identified in the CSF are not present in the serum. Therefore, a paired serum sample should be taken at the time of the lumbar puncture. They are identified in up to 95% of patients with definite MS clinically, but the test is only needed as supporting evidence in equivocal cases.
Typically, they can be used in patients with CIS as a surrogate markers of dissemination in time or as a risk estimation for those not yet meeting criteria for dissemination in space.
Visual evoked response (VER)
VER involves measuring electrical activity via a transducer over the occipital cortex in response to a light stimulus. It has limited sensitivity and specificity as a diagnostic test for previous optic neuritis and is not useful during acute episodes.
VER may be used to assess evidence of previous, asymptomatic episodes of optic neuritis. Up to 90% of individuals will have a persistent abnormality on VER following an acute episode at one year.
Antibody testing
Useful in the work-up of MS if an alternative demyelinating condition is suspected (e.g. Neuromyelitis optica). Both AQP4 and myelin oligodendrocyte glycoprotein (MOG) antibodies are associated with NMOSD.
Management
MS management is broadly divided into general care, managing acute relapses and disease modifying therapies.
As discussed, a range of clinical manifestations may develop in MS. Therefore, management is guided at treating a range of complications including depression, fatigue, bladder/bowel dysfunction, spasticity and poor motor function.
All patients with MS should be under the care of a specialist in MS with access to a multidisciplinary MS team including a clinical nurse specialist.
General care
There are many aspects to the general care of patients with MS. It should be emphasised that physiotherapy and occupational therapy are critical throughout the course of the disease, especially as the disease becomes more disabling and patients require assistance with their daily activities of living
- Bladder dysfunction: commonly seen in MS. May require anticholinergics such as oxybutynin for detrusor overactivity. Botulism injection is a second option if intolerant to medications. Patients with retention problems may require intermittent self-catheterisation or long-term catheters.
- Bowel dysfunction: constipation, poor evacuation and incontinence may be present. Dietary changes, laxatives and enemas are the cornerstone of treatment.
- Depression: treatment choices can be targeted depending on co-existing symptoms. For example, duloxetine may be preferred if co-existing neuropathic pain or fatigue. Selective serotonin reuptake inhibitors (SSRIs) commonly used.
- Fatigue: numerous non-pharmacological interventions should be attempted (e.g. physical activity) and treat co-morbidity such as depression. Some pharmacological options available like modafinil.
- Gait impairment: occupational and physiotherapy input essential. Walking aids or wheelchair may be needed.
- Pain: neuropathic pain significant issue in MS. Treatment options include amitriptyline, gabapentin or pregabalin.
- Spasticity: affects a large proportion of patients with MS. Can lead to functional disability. Degree of spasticity should be assessed and treatment involves physiotherapy and baclofen. Botulism injections may be used.
Many other aspects of care are essential in MS. These include respiratory function, swallowing and speech, vertigo, visual disturbances, seizures and sexual dysfunction. This highlights the wide-ranging effects of MS and need for a truly multidisciplinary team input.
Acute relapse
The principle management of an acute relapse or ‘attack’ in RRMS is the use of corticosteroids.
Before treating a relapse, it is essential to rule out infection, which can lead to a deterioration in symptoms. Furthermore, a true relapse must be distinguished from fluctuations in disease course or progressive disease. Patients may require hospital admission and should always be discussed with their specialist MS team.
If a clinically significant relapse is confirmed:
- Steroids: oral methylprednisolone 0.5 g daily for five days, OR Intravenous methylprednisolone 1 g daily for 3-5 days
- Gastroprotection: proton pump inhibitor
- Discuss risk/benefit of treatment: particular attention to acute changes in blood glucose and mental health
NOTE: mild relapses may not require steroid therapy.
Following treatment, recovery may be expected within 2-3 months in most cases, but there can be improvement up to 12 months. If the relapse is severe, more likely to be a residual functional disability.
Disease-modifying therapies (DMTs)
DMTs refer to immunomodulatory medications that aim to decrease the number of relapses and slow disease progression. The initiation of DMTs and ongoing care of patients on DMTs should be guided by MS specialists.
The initiation of DMTs have specific indications, but in general, the following principles should apply:
- No evidence of non-relapsing progressive MS
- Sustained disability due to MS: measured on Expanded Disability Status Scale (EDSS). The score should be < 7.0 (i.e. at least ambulant with two crutches).
DMTs utilised in MS:
- Interferon beta: injectable cytokine. Modulates immune response. First disease-modifying drug licensed for the treatment of MS.
- Glatiramer acetate: injectable. Mechanism not fully understood. Thought to induce and activate T-lymphocyte suppressor cells for a myelin antigen
- Teriflunomide: oral therapy. Inhibits pyrimidine synthesis, resulting in antiproliferative and anti-inflammatory effects
- Alemtuzumab: infusion. Monoclonal antibody targeted to CD52. Antigen is found on many immune cell lines. Immunomodulatory effect in MS.
- Cladribine: oral therapy. Purine nucleoside analogue. Cytotoxic effects on B and T lymphocytes through preventing DNA synthesis.
- Natalizumab: infusion. Monoclonal antibody against the alpha-4 subunit of integrin molecules. Prevents migration of leucocytes across the BBB.
There are various indications for the use of DMTs within MS. DMTs can be broadly divided into first-line, second-line and third-line treatment options. Most commonly, patients with RRMS and two clinically significant relapses over the last two years may be initiated on first-line DMTs (e.g. Interferon beta, Teriflunomide or Alemtuzumab).
If the disease remains active on first-line therapy, they can be switched to second-line therapy and then third-line therapy as necessary.
NOTE: further recommendations on the use of DMTs in MS is beyond the scope of these notes.
Prognosis
Prognosis in MS is highly variable between patients and ability to accurately predict outcomes is limited.
In general, 25% of patients with RRMS develop SPMS within six years of diagnosis and 50% within 15 years. Following diagnosis the likelihood of walking unaided after 15 years was 60%. In patients with PPMS the median time between disease onset and requiring a waking aid was eight years in one study.
Factors linked to prognosis include:
- Disease type: RRMS better prognosis than PPMS. Most patients with RRMS eventually develop SPMS.
- Recovery following first attack: incomplete recovery associated with worse prognosis.
- Clinical manifestations at onset: pyramidal, brainstem, and cerebellar symptoms (poor prognosis). Sensory symptoms, optic neuritis (favourable prognosis).
- Pregnancy: protective during pregnancy. Increased risk of relapse in postpartum period.
- Imaging: lesion load and cerebral atrophy linked to prognosis (higher burden and increased atrophy have worse prognosis).

